

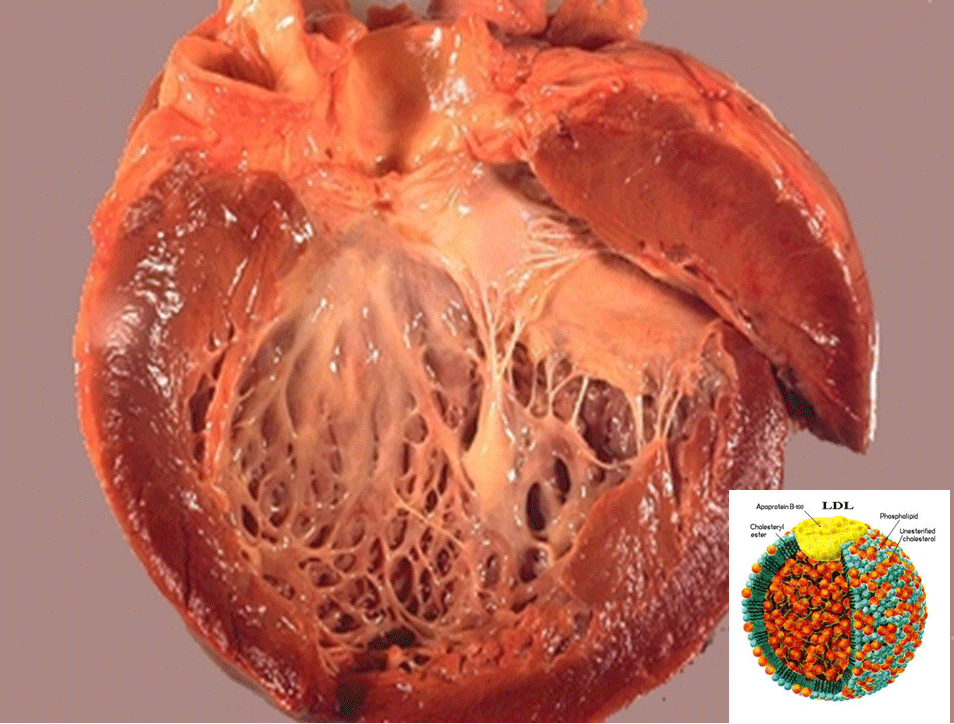
La cardiomiopatia aritmogena (ACM) è una malattia genetica in cui il miocardio ventricolare viene progressivamente sostituito da tessuto fibro-adiposo.
Si associa ad aritmie ventricolari gravi, morte improvvisa (soprattutto in giovani atleti), e insufficienza cardiaca progressiva.
In Italia la malattia è più diffusa che nel resto del mondo, con un picco di prevalenza nel Nord-Est, dove ne è colpita una persona su 2.000, contro un dato mondiale che registra un caso di malattia ogni 5.000-10.000 persone.
La sostituzione fibro-adiposa ha un doppio effetto negativo sul miocardio:
- peggiora l’aritmogenesi creando un substrato non conduttivo,
- causa una disfunzione ventricolare che porta a insufficienza cardiaca.
Sono stati individuati diversi geni associati alla malattia (per esempio il gene che codifica per la proteina placofilina PKP2), ma la variabilità nella penetranza ed espressività di tali geni suggerisce che accanto ai fattori genetici vi siano anche fattori ambientali (epigenetica).
Attualmente purtroppo non vi sono farmaci né terapie per la cardiomiopatia aritmogena. Nell’ambito di un progetto sono stati osservati, – in una coorte di 16 pazienti con ACM, – livelli plasmatici di colesterolo totale, di c-LDL e di LDL ossidate leggermente superiori ai controlli sani (rispettivamente: 211,3 ± 8,0 vs 185,0 ± 6.8 mg/dl e 133.4 ± 30.8 vs 111.1 ± 20.7 mg/dl), ipotizzando che le dislipidemie e lo stress ossidativo indotto da esercizio fisico possano rappresentare cofattori per la genesi di questa patologia.
Sono stati utilizzati colture di cellule stromali mesenchimali (MSC) per le indagini in vitro e modelli animali knok-out per il gene che codifica per la PKP2 per gli esperimenti in vivo.
Fornendo acidi grassi 13-idrossioctadecanoici ossidati (presenti nelle LDL ossidate) alle colture di cellule MSC di ACM, l’accumulo di lipidi aumenta, effetto che non si verifica in cellule non-ACM. Un effetto opposto è stato registrato somministrando antiossidante N-acetilcisteina.
Sono stati anche osservati l’attivazione trascrizionale del gene PPARg (codificante per una proteina che regola la differenziazione degli adipociti) e l’aumento dell’espressione di CD36 (una proteina con alta affinità per gli acidi grassi).
In vivo, sebbene i modelli animali non accumulassero spontaneamente adipociti nel muscolo cardiaco, una sostituzione fibro-adiposa è stato osservato dopo 3 mesi di dieta iperlipidica. Inoltre, le cellule stromali mesenchimali ottenute dai ratti knok-out per il PKP2, esposti a un mezzo di coltura adipogenico, accumulano lipidi, ciò che non avviene nei ratti wild-type.
In conclusione le mutazioni nei geni ACM sono necessarie ma non sufficienti per il manifestarsi della malattia e che elevati di LDL e stress ossidativo possono essere cofattori importanti.
CALABRESE Michele
Fonte:
Arnaboldi L – Experimental Cadiovascular Medicine II 2016; 252:e227–e228; Sommariva E – Eur Heart J. 2016;37(23):1835-1846.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lascia un commento