

La malattia di Gaucher è una malattia metabolica ereditaria rara determinata da un’alterazione del DNA che dà luogo a diverse mutazioni del gene GBA1.
Tali mutazioni causano l’assenza, o il non corretto funzionamento, dell’enzima (molecola che ha il compito di accelerare e facilitare le reazioni chimiche) chiamato “glucocerebrosidasi”. Esistono anche rari casi di malattia di Gaucher dovuti a deficit della saposina C, attivatore dell’enzima glucocerebrosidasi.
L’enzima glucocerebrosidasi degrada il glucocerebroside, un grasso che costituisce la membrana delle cellule. Quando l’enzima non funziona o non viene prodotto si verifica un accumulo di sostanze grasse in molti organi, come milza, fegato, cervello, muscoli, ossa, midollo osseo, ecc. Tale accumulo altera la normale struttura degli organi non consentendogli di funzionare come dovrebbero.
Il quadro clinico della malattia di Gaucher è molto vario, probabilmente in base alla mutazione del gene. Si distinguono 3 varianti principali:
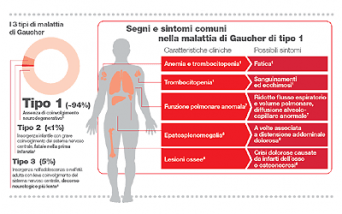
- tipo 1, è la più frequente, rappresenta il 95% dei casi ed è la forma cronica e non neurologica dell’adulto
- tipo 2, forma neuropatica acuta
- tipo 3, forma neuropatica cronica
I sintomi tipici della malattia possono comparire a qualsiasi età in numero e con gravità diversi da persona a persona. Il trattamento, di solito, prevede una terapia enzimatica sostitutiva (in sigla ERT, dall’inglese Enzyme Replacement Therapy) o altre terapie che arrestano, o rallentano, la progressione della malattia, migliorando la qualità della vita delle persone con malattia di Gaucher.
L’incidenza della malattia nella popolazione generale è circa 1:60.000, ma tra gli ebrei Ashkenaziti è più alta (1/1.000).
SINTOMI
I segni e i sintomi della malattia di Gaucher possono variare ampiamente da persona a persona, anche all’interno della stessa famiglia o tra fratelli gemelli identici.
Nella maggior parte dei casi compaiono:
- disturbi addominali, dolore all’addome causato dall’ingrossamento della milza (detto splenomegalia) e del fegato (detto epatomegalia); fitte al fianco durante l’attività fisica e il gioco, dolore nella parte alta dell’addome e diarrea
- anomalie scheletriche, la malattia di Gaucher può indebolire le ossa (si parla di osteopenia/osteoporosi), aumentando il rischio di fratture dolorose, anche spontanee. Può causare deformità a fiasca delle ossa e interferire con l’apporto di sangue al loro interno, causando edema ossei (o infarti ossei) e necrosi di porzioni di osso. Nei bambini è molto comune il ritardo della crescita scheletrica
- disturbi a carico del sangue, una diminuzione del numero di globuli rossi (detta anemia) può provocare senso di affaticamento; una riduzione dei globuli bianchi (detta neutropenia) può aumentare il rischio di infezioni. La malattia di Gaucher colpisce anche le piastrine, cellule responsabili della coagulazione del sangue, facendo diminuire il loro numero (si parla di piastrinopenia) con conseguente facilità alle emorragie, in particolare frequenti lividi e perdita di sangue dal naso
- disturbi a carico dei polmoni, iniziale mancanza di fiato durante lo sforzo e tosse causati dalla fibrosi pomonare che si determina per infiltrazione cellulare anomala. Successivamente, può comparire pressione alta nei vasi polmonari (si parla di ipertensione polmonare) con cianosi (ossia colorazione blu delle mucose ) e difficoltà di respirazione a riposo. L’insufficienza respiratoria è una grave conseguenza della malattia di Gaucher
- disturbi a carico del sistema nervoso, presenti nella malattia di Gaucher tipo 2 e tipo 3, sono per lo più rappresentati da movimenti oculari anomali, strabismo, rigidità muscolare (detta spasticità), difficoltà di deglutizione e convulsioni. La malattia di Gaucher tipo 2 è molto rara, inizia già nella prima infanzia e, in genere, porta a morte entro i primi tre anni di vita
- disturbi a carico di pelle, cuore, occhi e sistema linfatico, sono meno frequenti e comprendono, rispettivamente, alterazioni del colorito (giallo/olivastro), cardiomiopatia restrittiva, opacità della cornea e linfonodi ingrossati
CAUSE
La malattia di Gaucher è una malattia metabolica ereditaria causata da mutazioni del gene GBA1, localizzato nel cromosoma 1.
Si tratta di una malattia autosomica recessiva, cioè si verifica quando ambedue i genitori trasmettono ai figli il gene difettoso. La persona che eredita da un genitore una sola copia del gene malato è detto eterozigote o portatore sano della malattia di Gaucher. Ciò significa che il portatore sano non la svilupperà ma potrebbe trasmetterla ai figli.
Sono state identificate più di 300 mutazioni del gene GBA1, responsabili dell’assenza o della riduzione della quantità della proteina enzimatica glucocerebrosidasi. Come conseguenza di queste mutazioni, si verifica un accumulo di grassi (chiamati glicosfingolipidi) nei lisosomi, organelli simili a vescicole che si trovano all’interno delle cellule di tutti i tessuti dell’organismo.
DIAGNOSI
Si distinguono 3 tipi principali di malattia di Gaucher:
- tipo 1, è la forma cronica e non neurologica dell’adulto, rappresenta il 95% dei casi. Si tratta di una malattia prevalentemente caratterizzata da ingrossamento del fegato e della milza (si parla di epatosplenomegalia), anomalie scheletriche (ossia dolore, osteopenia/osteoporosi, infarti ossei, necrosi), disturbi a carico del sangue (riduzione del numero di piastrine e di globuli rossi e, raramente, diminuzione dei globuli bianchi) e coinvolgimento di altri organi
- tipo 2, è la forma neurologica infantile acuta. È caratterizzata da un prevalente coinvolgimento del sistema nervoso a causa di una grave disfunzione del tronco cerebrale. Compare durante il primo anno di vita, l’evoluzione è rapida e porta a morte entro i primi tre anni di vita
- tipo 3, è la forma neurologica subacuta, giovanile. È caratterizzata da danni progressivi al sistema nervoso (quali disturbi oculari, convulsioni e mancanza di coordinazione dei movimenti muscolari volontari) associati ai disturbi presenti anche nella malattia tipo 1. Compare nell’infanzia o nell’adolescenza
- tipo II pre-natale, in letteratura esistono rarissimi casi di segnalazioni del cosiddetto bambino “cellophane”, per l’aspetto liscio e teso della pelle. La morte avviene in utero o entro pochi giorni dalla nascita
I sintomi causati dalla malattia di Gaucher tipo 1 (e tipo 3, fino a quando non compaiono i sintomi e i segni neurologici) sono molto variabili e molto spesso non consentono al medico specialista di accertare la malattia in tempi brevi.
Ad esempio, l’osteoporosi o le fratture dolorose spingono la persona con malattia di Gaucher a consultare l’ortopedico, mentre l’anemia, la piastrinopenia e i dolori ossei a contattare un ematologo o un oncologo.
A volte, la diagnosi della malattia di Gaucher è casuale e si basa sull’esame istologico della milza prelevata chirurgicamente per scoprire le cause del suo ingrossamento (detto splenomegalia).
La diagnosi della malattia di Gaucher di tipo 2 è più facile sia per la presenza simultanea di disturbi neurologici associati all’ingrossamento di fegato e milza, sia perché i pediatri sono, in genere, più informati su questa malattia degli specialisti dell’adulto.
Per confermare la presenza della malattia di Gaucher si esegue anche il prelievo di una piccola quantità di sangue, da una vena del braccio, per verificare la quantità dell’enzima glucocerebrosidasi ed eseguire l’analisi genetica del gene mutato.
La consulenza genetica è molto importante per conoscere le conseguenze della malattia, il rischio di comparsa all’interno della stessa famiglia, gli interventi utili per controllarla e prevenirla. La diagnosi prenatale può essere eseguita prelevando del liquido amniotico (alla 14°-16° settimana di gravidanza) o, in epoca precedente, sui villi coriali (alla 10°-11° settimana di gravidanza).
La diagnosi genetica, dati i tempi di esecuzione, i costi elevati e il livello di specializzazione e strumentazione richiesti, è effettuata nei casi in cui i risultati delle analisi enzimatiche confermano la presenza della malattia per identificare il tipo di mutazione genica presente.
Mentre la diagnosi di portatore sano è limitata ai gruppi familiari in cui già si conosce il tipo di mutazione.
TERAPIA
Esistono diversi trattamenti che possono aiutare, con beneficio variabile, a tenere sotto controllo i sintomi, prevenire danni irreversibili e migliorare la qualità della vita delle persone con malattia di Gaucher.
Farmaci
Molte persone con la malattia di Gaucher hanno mostrato dei miglioramenti dopo aver iniziato le seguenti terapie farmacologiche:
- terapia enzimatica sostitutiva (ERT), arresta e inverte la progressione della malattia, migliorando la qualità della vita delle persone con malattia di Gaucher. La terapia, a base dell’enzima carente, è somministrata in regime ambulatoriale per via endovenosa a intervalli di due settimane. Il farmaco (imiglucerasi, velaglucerasi e taliglucerasi), nel tempo, determina soprattutto una progressiva riduzione dell’ingrossamento dei visceri (detto visceromegalia) e dei disturbi a carico delle cellule del sangue. Occasionalmente alcune persone manifestano una reazione allergica o un’ipersensibilità alla ERT. È la terapia di prima scelta ed è indicata nelle persone con malattia di tipo 1 e tipo 3; in quest’ultima forma, tuttavia, la ERT non sembra prevenire la progressione del danno neurologico
- terapia basata sulla diminuzione del substrato, utilizza i farmaci miglustat e l’eliglustat che agiscono impedendo l’azione di un enzima detto, sintasi della glucosilceramide, in modo tale da ridurre l’accumulo di grassi (in particolare i glicosfingolipidi) nelle cellule. Nausea e diarrea sono effetti indesiderati comuni. Questi farmaci sono impiegati in persone non adatte a essere trattate con la terapia ERT e richiedono valutazioni specifiche per ogni paziente da parte del medico
- trattamento dell’osteoporosi, i farmaci utilizzati per contrastare l’osteoporosi possono aiutare a ricostruire l’osso indebolito dalla malattia di Gaucher
Chirurgia
Se i sintomi sono gravi e la terapia non è stata iniziata tempestivamente, potrebbero essere necessari:
- trapianto di midollo osseo, prevede la rimozione e sostituzione delle cellule del sangue danneggiate dalla malattia di Gaucher. Può correggere molti dei segni e dei sintomi causati dalla malattia
- asportazione della milza, prima della disponibilità della ERT, la rimozione della milza era un trattamento molto comune per la malattia di Gaucher. Attualmente, è utilizzata come ultima risorsa
COMPLICAZIONI
La malattia di Gaucher può aumentare il rischio di:
- ritardo di crescita, nei bambini
- problemi ginecologici e ostetrici
- morbo di Parkinson
- tumori, come il mieloma, la leucemia e il linfoma
In Italia, la malattia di Gaucher è inserita nell’elenco delle malattie rare esenti dal costo del ticket (Decreto Ministeriale n. 279 del 2001) con il codice RCG080.
Per informazioni sulla malattia di Gaucher è possibile rivolgersi al Telefono Verde Malattie Rare (TVMR) 800.89.69.49. Il numero è attivo dal lunedì al venerdì dalle 9,00 alle 13,00.
Redazione NurseTimes
Rimani aggiornato con Nurse Times, seguici su:
Telegram – https://t.me/NurseTimes_Channel
Instagram – https://www.instagram.com/nursetimes.it/
Facebook – https://www.facebook.com/NurseTimes. NT
Twitter – https://twitter.com/NurseTimes










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lascia un commento