

La malattia di Creutzfeldt-Jakob (MCJ) è una rara patologia degenerativa del sistema nervoso centrale ad esito fatale
E’ caratterizzata da demenza rapidamente ingravescente e segni neurologici focali. Oltre alla MCJ appartengono alle Encefalopatie Spongiformi Trasmissibili (EST) la sindrome di Gerstmann-Sträussler-Scheinker (GSS), l’insonnia fatale familiare (FFI) e il kuru, una forma un tempo epidemica tra gli indigeni di lingua Fore dell’isola di Papua-Nuova Guinea ed oggi quasi scomparsa.
Le EST dell’uomo e degli animali sono trasmissibili, mediante inoculazione di tessuto infetto, ad una grande varietà di animali di laboratorio (primati, felini, ruminanti, roditori) dopo un lungo periodo di incubazione che può durare da alcuni mesi a diversi anni.
L’evento patogenetico fondamentale delle EST è costituito dalla formazione e accumulo, a livello cerebrale, di una proteina amiloidea caratteristica denominata PrPsc (sc da scrapie, dove è stata per la prima volta isolata).
La MCJ può presentarsi in forma sporadica, familiare o come conseguenza di trasmissione accidentale (forma iatrogena) da uomo a uomo in seguito a procedure di carattere medico.
Il quadro clinico d’esordio è rappresentato in circa la metà dei casi da deterioramento mentale, nel 40% dei casi da segni neurologici a focolaio e, nel restante dei casi, da entrambe le manifestazioni cliniche.
Il deterioramento mentale può manifestarsi con disturbi cognitivi, soprattutto deficit mnesici, o con modificazioni comportamentali.
I segni neurologici d’esordio più frequenti sono di natura cerebellare, quali per esempio l’atassia della marcia ed i disturbi dell’equilibrio, o visivi, come il nistagmo, la diplopia e le allucinazioni visive.
La sopravvivenza media è di circa 4-5 mesi, ma alcuni pazienti possono rimanere in vita in uno stato semi-vegetativo per più di un anno.
La diagnosi di MCJ può essere realizzata saggiando:
- anomalie elettroencefalografiche,
- analisi del Liquor (in tal caso il liquido cefalorachidiano non contiene è cellule infiammatorie, né proteine prioniche, ma nel 33% dei casi si assiste ad un incremento delle proteine).
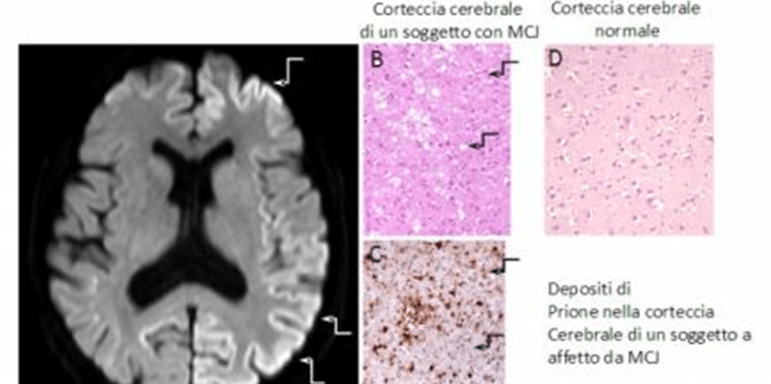
- La risonanza magnetica (RM) non mostr alterazioni focali franche, ma evidenzia segni di atrofia cerebrale con significative modificazioni del segnale e quindi della sua intensità a livello del nucleo caudato e putamen.
- La TAC mostra anch’essa segni di atrofia, ma spesso mostra immagini normali.
- PET e SPECT sono state impiegate per la diagnostica ma la loro utilità deve ancora essere validata dalla comunità scientifica.
- Esame neuropatologico: l’esame autoptico mostra con certezza, con l’ausilio dell’esame istologico, la presenza di malattia e la causa.
Un test, pubblicato sul New England Journal of Medicine,messo a punto dalla Clinica Neurologica di Verona, si esegue introdiucendo un tamponne nella cavitànasale fino al punto ove si localizzano i neuroni olfattori. Si esegue un prelievo di questo tessuto.
Un altro studio su Nature Communications ha dimostrato l’opportunità di diagnosticare la malattia di Creutzfeldt-Jakob analizzando la pelle di animali infetti. In sostanza Il gruppo di ricerca ha identificato la presenza della proteina prionica mediante due tecniche di isolamento della proteina PrPsc (RT– QuIC e la sPMCA). Successivamente sono stati prelevati campioni di cute in animali (criceto e topo transgenico) nei quali era stata inoculata la proteina prionica. Non solo le tecniche hanno dimostrato la presenza della proteina prionica a livello cutaneo, ma lo hanno anche fatto prima che fosse possibile riscontrarla a livello cerebrale e, conseguentemente, prima che i sintomi della malattia esordissero.
I promettenti risultati ottenuti nel modello animale aprono una strada alternativa per la diagnosi precoce della malattia di Creutzfeldt-Jakob.
Di seguito la classificazione dei tessuti che sono ad alto rischio di trasmissione della malattia
- distribuzione dei prioni nei tessuti*
- CATEGORIA I: alta infettività
- cervello
- midollo spinale
- occhi
- CATEGORIA II: media infettività
- sistema reticolo endoteliale
- tonsille
- linfonodi
- liquido cerebrospinale
- ileo, colon prossimale
- ghiandole surrenali
- ipofisi
- dura madre
- placenta
- CATEGORIA III: infettività non definibile
- muscoli
- cuore
- ossa
- ghiandola mammaria
- latte
- siero
- urine
- feci
- saliva
- e altri tessuti
- CATEGORIA I: alta infettività
* tratto da: rapporto dell’OMS sui medicinali e altri prodotti in relazione alle encefalopatie spongiformi trasmissibili umane e animali, Ginevra, 1997
CALABRESE Michele
Fonti
https://www.iss.it
https://www.salute.gov.it
https://www.msdmanuals.com










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lascia un commento