

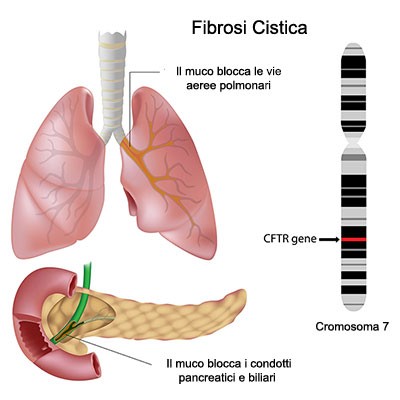
La fibrosi cistica è una malattia ereditaria grave, con il gene mutante responsabile della malattia, il CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), presente ereditato sia dal padre sia dalla madre. I genitori sono portatori sani
Il CFTR è una proteina deputata alla funzione secretoria di molti organi. Tale proteina funziona poco o per niente in chi nasce con la doppia copia del gene mutato. In Italia c’è un portatore sano ogni 25 persone circa.
La fibrosi cistica, quindi, altera le secrezioni di molti organi.
Le secrezioni solitamente possono presentarsi aumentate di densità, quantità o addirittura insufficienti per quella che è la fisiologia dell’organo interessato.
Generalmente gli organi che subiscono danni maggiori e che richiedono interventi di intensità assistenziale complessi sono i polmoni ed i bronchi.
Infatti qualora le secrezioni bronco-polmonari divenissero dense e difficili da espettorare, si crea terreno fertile per moltiplicazione batterica e infezioni.
A livello pancreatico si hanno alterazioni nell’escrezione di enzimi deputati alla digestione, con conseguente status di malassorbimento, ipermotilità intestinale, fino a deficit nutrizionali abbastanza importanti che potrebbero inficiare sulla crescita del paziente con Fibrosi Cistica.
Fino ad oggi le cure sono dirette ai sintomi e alla prevenzione delle complicanze, ma grazie alla ricerca si è giunti a delle molecole in grado di dare risposte incoraggianti nei confronti della malattia.
La triplice terapia con elexacaftor/tezacaftor/ivacaftor , secondo risultati di studi in fase III, e pubblicati su New England Journal of Medicine e su The Lancet, hanno sortito significativi miglioramenti della funzionalità polmonare e di altri indicatori della malattia.
I dati evidenziano gli endpoint primari e secondari di uno studio di fase III di 24 settimane condotto su 403 persone. Elemento positivo è anche il fatto che la triplice terapia è stata generalmente ben tollerata.
I risultati sono stati pubblicati online in contemporanea alla presentazione di entrambi gli studi alla 33a North American Cystic Fibrosis Conference (NACFC) annuale, svoltasi dal 31 Ottobre al 2 Novembre a Nashville 2019.
Attualmente l’analisi della commercializzazione nel nostro Continente è all’analisi dell’Agenzia europea per i medicinali.
CALABRESE Michele
Fonte







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lascia un commento