

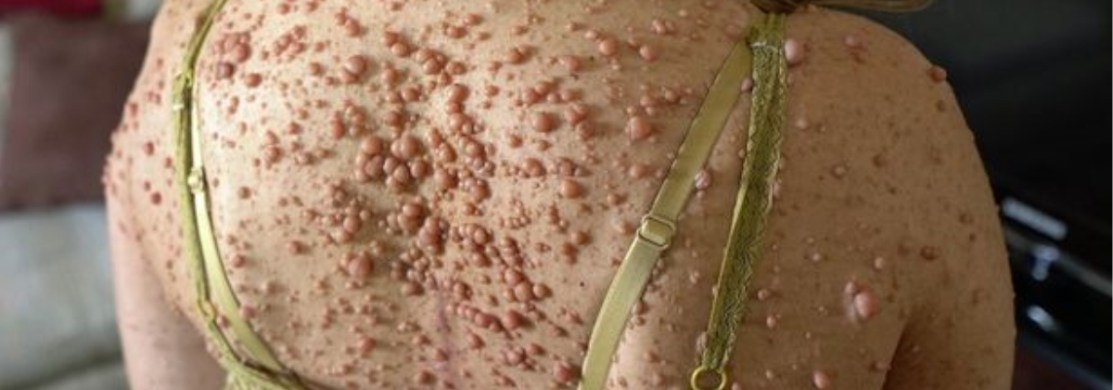
Le facomatosi sono patologie che interessano il sistema nervoso e la cute che talvolta possono colpire anche altri tessuti determinando danni a carico di altri organi e/o tessuti
Esse sono malattie genetiche che possono avere trasmissione dominante o recessiva anche se non sono escluse manifestazioni ex-novo della malattia.
Esempio di facomatosi sono le neurofibromatosi, diagnosi clinica e strumentale, e che provocano dei tumori che coinvolgono nervi centrali o periferici.
I tipi di tumore possono essere benigni o maligni che come manifestazione neurocutanea possono presentarsi con diverse modalità.
Clinicamente si presentano come macchie cutanee, manifestazioni tumorali da cellule embrionarie indifferenziate ed iperplasie di vario tipo.
Analizziamo due tipi di neurofibromatosi: tipo 1 e tipo 2.
Il tipo 1 colpisce una persona ogni 3.000, comunque, l’espressività della malattia è solitamente lieve con un limitato numero di tumori.
Invece, il tipo 2 è più grave ed interessa una persona ogni 50.000. Il tipo 2 causa multiple tumori del sistema nervoso centrale, mentre il tipo 1, che è la forma più comune, di solito causa tumori benigni fuori dal sistema nervoso centrale.
La neurofibromatosi di tipo 1 (NF1) o malattia di Von Recklinghausen (dal patologo tedesco Friedrich Daniel von Recklinghausen) ha lo spettro delle lesioni della malattia che variano ampiamente.
Sono presenti segni e sintomi di interessamento di più organi e tessuti.
Nel tipo 1 i segni clinici principali sono: macchie cutanee caffelatte, che sono presenti fin dalla nascita ed aumentano di numero con l’ età; neurofibromi periferici, che si evidenziano nel 50% dei pazienti tra i 10-20 anni e nel 70% dei pazienti oltre i 20 anni; noduli melanocitici (amartomi) dell’iride.
Il tipo 2 è caratterizzato da macchie caffelatte; opacità posteriore del cristallino, che causa disturbi dell’acuità visiva; neurofibromi periferici; neurinoma acustico, bilaterale nel 95% dei casi, e tumori del sistema nervoso centrale.
La sintomatologia dipende dalla sede del tumore, possono essere presenti ipertensione endocranica, ipoacusia progressiva, acufeni, disturbi dell’ equilibrio, ecc.
La diagnosi di neurofibromatosi tipo 1 è possibile se sono presenti almeno due dei seguenti criteri:
- sei o più macchie caffellatte (discromia bruna della cute) con diametro maggiore di 5 mm in epoca prepuberale, e maggiore di 15 mm in epoca postpuberale;
- due o più neurofibromi;
- iperpigmentazione rossastra delle regioni ascellari o inguinali;
- glioma del nervo ottico;
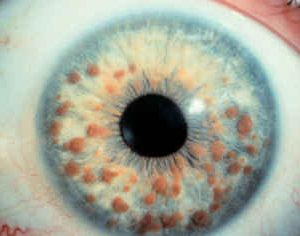
- due o più noduli di Lisch (piccole masse sull’ iride, visibili alla lampada a fessura);
- anomalie ossee (displasia osso sfenoidale, cifoscoliosi e cisti ossee);
- un parente di primo grado (genitore, figlio o fratello) con neurofibromatosi di tipo 1.
I criteri diagnostici per il tipo 2 richiedono: neurinoma dell’ottavo nervo cranico, bilaterale; oppure un parente di primo grado con neurofibromatosi 2 e un neurinoma unilaterale dell’ottavo o due delle seguenti lesioni: neurofibroma, meningioma, glioma, schwannoma (classificata come neurofibromatosi di 3° tipo).
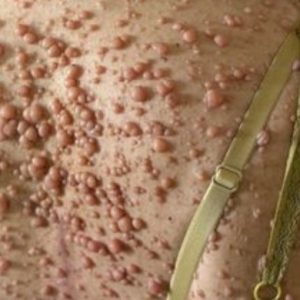
La diagnostica per immagini si avvale di Risonanza Magnetica o della radiologia tradizionale per l’analisi delle singolarità patognomoniche ossee extracraniche
La chirurgia è indicata per evitare la compressione da parte delle masse tumorali di organi o strutture anatomiche delle quali pregiudicano funzionamento.

Infatti la prognosi della malattia è funzionale alla qualità e quantità delle lesioni presenti in un determinato distretto.
CALABRESE Michele
Fonte:







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lascia un commento