


Le malattie delle ghiandole endocrine rappresentano condizioni patologiche che coinvolgono principalmente, ma non sempre esclusivamente, il sistema endocrino, costituito da ghiandole che producono sostanze chiamate ormoni.
Il normale funzionamento delle ghiandole distribuite nei diversi distretti corporei (cervello, apparato digerente, rene, surrene, organi genitali, tessuto adiposo, cute, eccetera) garantisce normali livelli di ormoni secreti e un regolare equilibrio delle funzioni vitali dell’organismo. Alcune malattie endocrine sono rare, colpendo relativamente pochi individui nella popolazione generale. In questo articolo daremo uno sguardo alla Sindrome di Prader-Willi, una malattia genetica di tipo endocrino-metabolico, caratterizzata da anomalie a livello dell’ipotalamo e molto rilevante in campo clinico-assistenziale.
La sindrome di Prader-Willi è una malattia genetica rara (1 ogni 25.000-30.000 nuovi nati, colpendo in egual misura maschi e femmine senza alcuna predilezione per nessuno dei due generi), purtroppo ancora sottostimata anche nel nostro Paese a causa della scarsa conoscenza che si ha in essa, in considerazione del fatto che in Italia, infatti, dovrebbero esserci circa 3.000 casi, ma ne sono stati riconosciuti ufficialmente “solo” 600.
Un bambino affetto da Prader-Willi è privo del senso di sazietà, condizione dovuta alle anomalie ipotalamico-pituitarie che in condizioni fisiologiche, tra le altre cose, garantisce una corretta regolazione del senso della fame e della sazietà.
Allo stesso tempo, la malattia è causa di una disfunzione nel metabolismo, che riduce notevolmente la capacità dell’organismo di bruciare calorie nutrienti assunti con l’ alimentazione. L’associazione di mancato stimolo della sazietà e ridotto metabolismo di grassi e calorie, spinge il bambino a raggiungere un peso corporeo eccessivamente elevato, fino a raggiungere persino i 200-300 kg di peso. È necessaria, quindi, un’informazione continua e capillare sull’ esistenza di questa malattia, permettendo una diagnosi precoce e un’ assistenza continua.
Conosciuta nel 1956, la patologia è dovuta ad una mutazione del cromosoma 15, anche se non è ancora chiaro quali geni siano esattamente coinvolti, anche se recenti ricerche puntano a capire come la metilazione distingue gli alleli materni e paterni.
I geni SNRPN, MKRN3 e NDN sono stati individuati e studiati ed è risultato che siano espressi solo dall’allele ereditato dal padre, e quindi i pazienti con Sindrome di Prader-Willi non esprimono tali geni. Tutti questi geni hanno un’isola CpG al 5′, che non è metilata nell’allele paterno espresso, mentre viene metilata in quello sotto imprinting (materno). Una scoperta importante è che il segnale di imprinting – fenomeno fisiologico delle cellule somatiche – per il gene SNRP inizia già nella gametogenesi maschile e femminile.
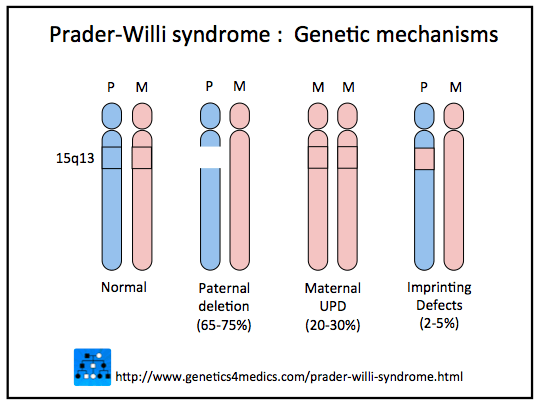
Mediante test genetici, si è potuto appurare che i pazienti con sindrome di Prader-Willi mancano di un cromosoma 15 paterno normale o comunque ad anomalie della regione critica del cromosoma 15 (15q11-q13). Ciò è deleterio nell’ipotalamo, dove l’unica linea cromosomica attiva è quella paterna.
Gli esperti quindi concordano che la diagnosi debba basarsi su criteri clinici (i criteri di Holm del 1993, rivisti nel 2001) ed essere confermata dalle analisi genetiche, che trova nella metodica più specifica il Test di Metilazione.
Attraverso questo test viene effettuato un Southern Blot che consiste nell’ibridare il DNA del paziente (dopo trattamento con enzimi di restrizione) con DNA “sonda” marcato nelle zone critiche da evidenziare. Se l’ibridazione (ovvero l’appaiamento tra il DNA sonda e quello del paziente) è pressoché perfetta, il paziente non è affetto dalla patologia; se invece presenta appaiamenti erronei sarà la prova significativa che nel paziente vi sono mutazioni geniche.
Questa tecnica non distingue le varie mutazioni, ma rappresenta la tecnica d’elezione perché più economica di altre e permette di diagnosticare/escludere la PWS (Prader Willi Syndrome) al 100%.
La maggior parte dei casi è sporadica e i casi familiari sono rari; tutte queste informazioni dovrebbero essere fornite aldilà di tutto dalla consulenza genetica.
Le ragioni possibili del malfunzionamento o dell’assenza del cromosoma 15 paterno sono tre:
- Assenza di una regione specifica del cromosoma 15 paterno, il che significa che il cromosoma manca di una parte essenziale. Rappresenta la maggior parte del meccanismo comune (65-75%).
- Presenza di due cromosomi 15 materni, un anomalia causata da un errore durante la formazione dell’embrione. Rappresenta il 20-30% dei casi.
- Mutamento di alcuni geni presenti sul cromosoma 15 paterno. Molto raro (2-5% dei casi), per lo più si verifica a causa di mutazioni epigenetiche e in alcuni sono dovuti a microdelezioni nella regione ipotalamica che vedono, in tal caso, un rischio di recidiva del 50 %.
La Sindrome di Prader-Willi si manifesta, con i primi sintomi e segni, già durante il primo anno di vita. In questo periodo, provoca principalmente una riduzione del tono muscolare (ipotonia) e un ritardo dello sviluppo, oltre che spossatezza, difficoltà del bambino nel svegliarsi ed una facies caratteristica (occhi a mandorla, restringimento della testa in corrispondenza delle tempie, bocca rivolta verso il basso e labbro superiore sottile).
Dopo il primo anno di vita insorgono generalmente una lunga serie di problematiche che possono avere esiti drammatici, e che riguardano:
- Iperfagia e obesità; i pazienti mostrano una costante voglia di cibo, che li porta a mangiare tantissimo e ad ingrassare notevolmente non provando il cosìddetto senso della sazietà. Se non trovano di che alimentarsi, arrivano a consumare cibi congelati e rifiuti, in altre parole qualsiasi cosa di edibile. Tutto ciò si deve alle alterate funzioni dell’ipotalamo che stanno all’origine della malattia.
- Ipogonadismo; condizione in cui gli organi genitali (testicoli e ovaie) producono pochi ormoni sessuali (testosterone ed estrogeni). Il paziente pertanto non riesce a completare lo sviluppo puberale fisiologico, e presenterà quasi sempre infertilità.
- Crescita e sviluppo ridotto; Infatti il paziente affetto da Prader-Willi raggiungerà mediamente un altezza che oscilla tra un 1.45 – 1.55 cm.
- Deficit di apprendimento; riduzione delle facoltà intellettive sono quasi sempre presenti.
- Ritardo motorio; i bambini imparano a camminare molto tardi.
- Disturbi del sonno; ricordando che il ciclo sonno-veglia è controllato dall’ipotalamo, i pazienti affetti da Prader-Willi presentano difficoltà nell’alternare le fasi REM del sonno alle fasi NON-REM, dovuto proprio ai disturbi ipotalamici-pituitari che stanno alla base della patologia. Inoltre i pazienti quando dormono, soffrono di interruzioni del respiro (apnee del sonno).
- Problemi comportamentali; i bambini si presentano testardi, capricciosi con presenza del famoso disturbo ossessivo-compulsivo (cioè atti riguardanti comportamenti ripetitivi o azioni mentali che il soggetto non riesce a non fare e che hanno lo scopo di ridurre l’ansia o il disagio, o di prevenire gli eventi o le situazioni temute).
Proprio su quest’ultimo punto L’articolo di ricerca “Life Span and Disability / Ciclo Evolutivo e Disabilità / XIII, 2 (2010), 259-274 Autolesionismo: Confronto tra sindrome di Prader-Willi, sindrome di Down e Autismo” di Serafino Buono, Fabio Scannella & Maria B. Palmigiano, ha raccolto i risultati di uno studio condotto volto a rivelare la presenza di comportamenti autolesionisti in un campione composto da 81 persone affette da Sindrome di Prader Willi, Sindrome di Down, Disturbo autistico.
I comportamenti autolesionisti sono stati rilevati attraverso una Scheda di Rilevamento di comportamenti autolesivi (SRCA) da parte di psicologi ai familiari e ai caregiver dei pazienti nel reparto di riabilitazione e al servizio di diagnostica dell’IRCSS Oasi Maria SS di Troina (EN).
Le schede permettevano di rilevare l’età di insorgenza dei SIB (Self Injurious Behavior ovvero “comportamento di autoferimento” ) e le zone corporee principalmente colpite. Dall’analisi delle schede di rilevamento, è emerso che l’esordio dei SIB avviene prevalentemente durante la prima infanzia (<7 anni) in tutte e tre le patologie, ma nella PWS avviene nel 90% dei casi. Sempre rimanendo nel gruppo di persone esaminate con la PWS, le aree prevalentemente colpite sono le mani (90%), le braccia (70%), la bocca e la testa (30%), il naso, le guance e le orecchie (20%). I comportamenti frequentemente rilevati sono per la SPW risultano essere lo skin-picking (50%), lo strapparsi/mangiarsi le unghie (40%), lo strapparsi i capelli (30%), il graffiare ed inserire dita/oggetti nelle cavità (20%).
Per i familiari e i caregiver è necessario conoscere tutte le corrette prassi da attuare per la gestione dei comportamenti che la patologia comporta. Nel caso di comportamenti lesivi bisogna attuare strategie tali da ammortizzare/eliminare codesti modi di agire, come ad esempio tenendo occupato il bambino con giocattoli per distogliere l’attenzione dal comportamento compulsivo che il soggetto presenta nell’auto-infliggersi un danno; promettere una ricompensa qualora non si “stuzzichi” o maltratti nessuna parte del corpo, o quando la ferita si rimargina; osservare attentamente la frequenza degli eventi autolesivi poiché essi sono indice di elevati livelli di ansia che sfociano in questi comportamenti ossessivo-compulsivi.
Essendo un’affezione genetica non esiste purtroppo una terapia farmacologica capace di debellare la malattia. La somministrazione di ormoni sessuali (testosterone per l’uomo ed estrogeni per la donna) risulta essere fondamentale per ripristinare i livelli ridotti di questi 2 ormoni, i quali oltre a migliorare la fertilità, possono riuscire a contrastare anche l’osteoporosi. I pazienti con Sindrome di Prader-Willi necessitano di una riabilitazione fisica e del linguaggio, attraverso l’intervento di professionisti della salute quali fisioterapisti e logopedisti.
La prima mira a limitare l’ipotonia muscolare e gli effetti dell’obesità; la seconda rimedia alle carenze comunicative, sia verbali che non verbali.
Per quanto concerne la psicoterapia, essa è fondamentale per quei pazienti con disturbi ossessivi-compulsivi e dell’umore in generale. Il supporto di uno psichiatra o di uno psicologo può migliorare notevolmente l’aspetto comportamentale
La terapia occupazionale, invece, ha l’obiettivo di insegnare al malato come prendersi cura di sé, come vestirsi ecc., in altre parole come svolgere le principali attività quotidiane. Ma in tutti questi rimedi terapeutici, bisogna dire come risulti essere di fondamentale importanza il supporto dei familiari nei confronti del paziente, specie durante gli anni della gioventù.
In ambedue le circostanze risulta essere necessario l’intervento dell’infermiere, attraverso un approccio assistenziale di elevata caratura. I consigli che il professionista infermiere deve dare solitamente alle famiglie sono molteplici. Egli infatti ha il compito di istruire la famiglia nel seguire il paziente in ogni sua attività (in particolare quando questo si alimenta); deve dare informazioni sui comportamenti più appropriati da riservargli, facendolo sentire sempre partecipe senza mai escluderlo; insegnare ad educarlo all’ascolto, al dialogo, al mantenimento delle funzioni cognitive/motorie ecc.
Nel campo della prevenzione della malattia, sono poche sono le manovre attuabili per anticipare l’insorgenza della malattia. Quando la Sindrome insorge nell’embrione a causa di una mutazione genetica, non vi è modo di prevenirla.
Tuttavia se, invece, due genitori hanno già dato alla luce un bambino con Sindrome di Prader-Willi, prima di una seconda gravidanza possono sottoporsi a determinati test genetici per scoprire se sono portatori o meno della malattia.
Infine per quanto concerne le considerazioni sulla qualità della vita, la salute generale del soggetto con Sindrome di Prader-Willi viene considerata di solito buona dalla PWSA (Prader Willi Syndrome Association) degli USA, con ottimismo, ma anche in modo obiettivo. Se il peso è sotto controllo, le aspettative di vita possono essere normali.
La costante necessità di restrizione del cibo e di controllo del comportamento deve essere chiaramente presente in ciascun familiare di soggetto con PWS. Adolescenti ed adulti possono trarre vantaggio da programmi di vita gruppo organizzata, sempre che vi sia un rigido controllo della dieta ed un ambiente adeguato.
Una patologia che sembra non lasciare “scampo” per il paziente, sopratutto viste le enormi difficoltà che il soggetto vive, in costante lotta con il suo desiderio di migliorare la sua condizione (specie nell’alimentarsi) che non riesce a controllare nonostante la sua forza di volontà e che sfociano spesso in esternazioni rabbiose ed aggressive che sicuramente possono essere controllate ed attenuate nelle manifestazioni della sintomatologia caratteristica come abbiamo visto attraverso una terapia sia farmacologica che sopratutto psico-assistenziale.
Quest’ultimo approccio diventa fondamentale per regalare una vita più dignitosa possibile sia ai pazienti affetti che purtroppo non riescono a controllare i propri sintomi pur mostrando coscienza e tenacia da vendere, sia ai familiari, in cui risulta essere fondamentale la consulenza infermieristica, mirata a porvi le corrette norme di educazione sanitaria, attraverso un intervento multidisciplinare che vede la collaborazione tra molte figure medico-sanitarie già citate, affinchè si possano combattere queste “enormi battaglie” che questa Sindrome purtroppo investe nel contesto familiare, con interventi terapeutici-educazionali mirati e costanti e che magari possono ritornare utilissimi a non far “perdere la guerra” …!
Per maggiori informazioni inerenti la Sindrome di Prader-Willi, consultate il sito web: www.praderwilli.it
Gaetano Ciscardi
SITOGRAFIA
- www.palidoronews.it
- mgm.ufl.edu
- it.wikipedia.org
- genetics4medics.com
- www.my-personaltrainer.it
- www.fmag.unict.it
- www.amaram.it











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lascia un commento